Reação de Wittig
A reação de Wittig é uma reação química de um aldeído ou uma cetona com um ileto de fósforo para dar um alceno.[1][2]

Foi descoberta em 1954 por Georg Wittig. Por este motivo lhe foi concedido o Prêmio Nobel de Química em 1979. Se usa amplamente em síntese orgânica na preparação de alcenos.
Mecanismo de reação
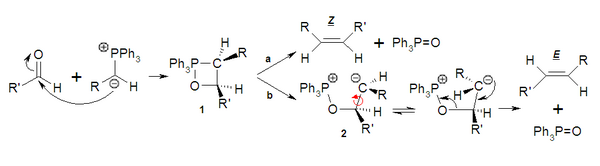
O carbono carregado negativamente do ileto de fósforo (1) pode atuar como nucleófilo e atacar o carbonilo (2). Isto conduz à betaína (3), que é uma espécie de tipo zwitterion. Esta forma rapidamente o heterociclo sem separação de cargas dando lugar ao oxafosfetano (4). Até que ponto a reação em continuação evolua de forma concertada (a) ou via um intermediário (b) mediante a forma zwitteriônica (5) condicionará a estereoseletividade final. A estabilidade do óxido de trifenilfosfina (7) impulsiona a reação até os produtos obtendo-se o alceno (6).

Preparação dos iletos de fósforo
Os iletos (espécies que, no seu estado fundamental, têm cargas opostas em atomos adjacentes e cujo carbono é o mais electronegativo[3]) de fósforo se preparam a partir de haloalcanos e trifenilfosfina. Em uma primeira etapa se produz a substituição nucleofílica do haleto pela trifenilfosfina para dar lugar a um sal de fosfônio:
- Ph3P + X−CH2R → [Ph3P+−CH2R]X−
A carga positiva sobre o átomo de fósforo provoca uma certa acidez nos hidrogênios do carbono contíguo. Na continuação se gera o ileto de fósforo, também chamado fosforano, usando uma base, tal como alcóxidos, hidreto de sódio, butil-lítio, fenil-lítio etc. Meios de reação habituais são éteres como o tetraidrofurano (THF), o éter dietílico ou o éter dimetílico (DME). Normalmente os iletos se geran in situ.
- [Ph3PCH2R]X + B- → Ph3P=CHR + HB + X-
O grupo R determina a natureza, a força da base a empregar. Como veremos, R também influirá na estereoquímica do produto da reação de Wittig.
Iletos estáveis e inestáveis. Estereoseletividade
Sendo o sal de fosfônio [Ph3PCH2R]X então:
- Se o resíduo R é igual a H, alquila ou alcóxi, que não estabilizam a carga negativa sobre o carbono no ileto, se necessitam bases fortes como amideto de sódio (NaNH2), butil-lítio (BuLi), diisopropilamideto de lítio (LDA) ou hidreto de sódio (NaH) para gerar o ileto. Neste caso se diz que o ileto é instável, portanto muito reativo. A estereoquímica do produto é Z.
- Se R é um fenil (Ph) ou um vinil (−CH=CH2), que estabilizam por ressonância levemente a carga negativa, se necessitam bases menos fortes como podem ser alcóxidos tais como etóxido (EtO-) ou terc-butóxido (tBuO-). Estes iletos são "semi-estáveis". Neste caso a reação pode ser pouco estereoseletiva. O resultado é uma mescla de isômeros Z e E.
- Se R é uma carbonila, ciano (−CN) ou um éster (−COOR), capaz de estabilizar por ressonância a carga negativa, se podem usar bases mais débeis como NaOH, KOH, K2CO3. São conhecidos como iletos estáveis, são menos reativos que os anteriores. A reação é estereoseletiva dando produto E.
Assim pois (ainda que com bastante simplificação, já que a princípio se formam mesclas dos dois isômeros):
- - ileto estabilizado Alqueno trans ou E.
- - ileto não estabilizado Alqueno cis ou Z.

No caso que foram dois os substituintes unidos ao carbono ([Ph3PCHRR']X) que estabilizaram a carga negativa se chegaria à situação de iletos inertes, que não dão reação.
Causa da estereoseletividade
Quando o lieto ataca a carbonila se tem observado que a aproximação é sempre cis (1). Se R é um grupo que não estabiliza a carga negativa, como podem ser um H ou um alquilo, o oxafosfetano (1) de forma concertada (a) conduz ao produto, neste caso o alqueno cis ou Z. Em câmbio se R é um grupo atrator ou receptor de elétrons, capaz de estabilizar a carga negativa sobre o carbono, via b se forma um intermediário (2) que é uma forma zwitteriônica que tem um certo tempo de vida, o que lhe permite equilibrar até a forma mais estável, com menos impedimento estérico, dando lugar ao produto trans ou E.
Valor sintético
A reação de Wittig se tem convertido em um método popular de síntese de alquenos precisamente a causa de sua grande aplicabilidade. A diferença das reações de eliminação, a reação de Wittig forma a ligação dupla em uma posição totalmente determinada sem ambiguidade.
A reação de Wittig se pode levar a cabo na presença de funções alqueno, alquino, éster ou éter no composto carbonílico.
Dado que a reação funciona melhor com aldeídos que com cetonas, devido a que estas últimas são menos eletrófilas, o problema se agrava com cetonas impedidas estéricamente, onde a reação pode dar rendimentos pobres, em particular sobre tudo com iletos estabilizados. Em tais casos a alternativa é a reação de Horner-Wadsworth-Emmons (usando fosfonatos).
Modificação de Schlosser
Permite obter alquenos E com iletos não estabilizados,[4] a diferença da reação de Wittig clássica que conduz nestos casos ao isômero Z.

A baixa temperatura usando fenil-lítio (PhLi), que é uma base muito forte, se desprotona a betaína eritro (1), com os dois substituintes eclipsados, no carbono com o próton mais ácido, que é o contiguo ao átomo de fósforo carregado positivamente, dando lugar a um β-oxidoileto eritro (2). Aumentando suavemente a temperatura este se equilibra à configuração treo (3) mai estável, onde os dois grupos já não estão eclipsados e portanto o impedimento estérico é menor. O tratamento final com terc-butanol (tBuOH), ou outro ácool, acompanhado de um aumento de temperatura conduz ao isômero E.
Exemplos
Dada sua confiabilidade e versatilidade, a reação de Wittig se tem convertido em uma ferramenta habitual para os químicos orgânicos sintéticos.[5]
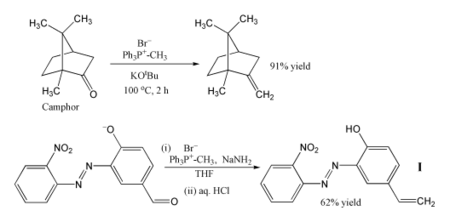
O uso mais popular da reação de Wittig é a introdução de um grupo metileno (=CH2) empregando trifenilmetilenofosforano (Ph3P=CH2). No exemplo mostrado, inclusive uma cetona impedida estericamente como o alcanfor pode ser convertida com exito a seu derivado metilênico aquecendo-a junto com brometo de trifenilmetilfosfônio e terc-butóxido de potássio, que generam o ileto in situ.[6] No outro exemplo, o fosforano (ou ileto) se produz usando amideto de sódio como base, convertendo-se eficientemente o aldeído mostrado no alqueno I com um rendimento de 62%.[7] A reação é levada a cabo em tetrahidrofurano (THF) frio, e os grupos nitro, azo e fenóxido permanecem intactos. O produto pode ser incorporado como fotoestabilizador em um polímero, para protegê-lo da radiação UV.
Outro exemplo de seu uso é na síntese do éster metílico do leucotrieno A.[8][9] O primeiro passo usa um ileto estabilizado, onde o grupo carbonilo está conjugado com o ileto prevenindo a autocondensação, ainda que inesperadamente isto conduz principalmente ao isômero cis. A segunda reação de Wittig utiliza um ilet não estabilizado, e tal como se esperava isto resulta principalmente no produto cis. destaca-se que as funções epóxido e éster permanecem intactas.

Em outro exemplo, o uso de um ileto funcionalizado como o trifenil-(metoximetileno)-fosforano (Ph3P=CHOMe) nos permite funcionalizar com um grupo formilo (-CHO) a posição carbonílica de um composto do tipo cetona (RR'C=O). Primeiro tem lugar a reação de Wittig formando-se o éter de enol (RR'C=CHOMe) correspondente. A continuação se leva a cabo a hidrólise com um ácido como catalisador, resultando o enol (RR'C=CHOH) que por tautomeria conduz ao aldeído (RR'CHCHO).
- RR'C=O + Ph3P=CHOMe → RR'C=CHOMe + Ph3P=O
- RR'C=COMe + H2O → RR'C=CHOH + MeOH
- RR'C=CHOH → RR'CHCHO
Referências
- ↑ Wittig, G.; Schöllkopf, U. Ber. 1954, 87, 1318
- ↑ Wittig, G.; Haag, W. Ber. 1955, 88, 1654
- ↑ P.Sykes, M.Sc., Ph.D., F.R.S.C., C.Chem. A Guidebook to Mechanism in Organic Chemistry, 5th Ed., Logman, UK, 1981, pag. 268
- ↑ Schlosser, M.; Christmann, K. F. Angew. Chem. Int. Ed. Engl. 1966, 5, 126.
- ↑ Maryanoff, B. E.; Reitz, A. B. Chem. Rev. 1989, 89, 863-927. (Review, doi:10.1021/cr00094a007)
- ↑ Fitjer, L.; Quabeck, U. Synthetic Communications 1985, 15(10), 855-864.
- ↑ Bottino, F. A.; Di Pasquale, G.; Pollicino, A.; Recca, A.; Clark, D. T. Macromolecules 1990, 23(10), 2662-2666.
- ↑ Ernest, I.; Main, A. J.; Menassé, R. Tetrahedron Lett. 1982, 23(2), 167-70.
- ↑ Corey, E. J.; Clark, D. A.; Goto, G.; Marfat, A.; Mioskowski, C.; Samuelsson, B.; Hammerström, S. J. Am. Chem. Soc. 1980, 102, 1436. (doi:10.1021/ja00524a045)
Bibliografia
- K. Peter C. Vollhardt (1994), Química Orgánica, Barcelona: Ediciones Omega S.A.. ISBN 84-282-0882-4.
- W.R. Peterson (1996), Formulación y nomenclatura química orgánica, Barcelona: EDUNSA - Ediciones y distribuciones universitarias S.A.. ISBN 84-85257-03-0.
 | Este artigo sobre Química é um esboço. Você pode ajudar a Wikipédia expandindo-o. |








